Photo-responsive monomers for ROMP
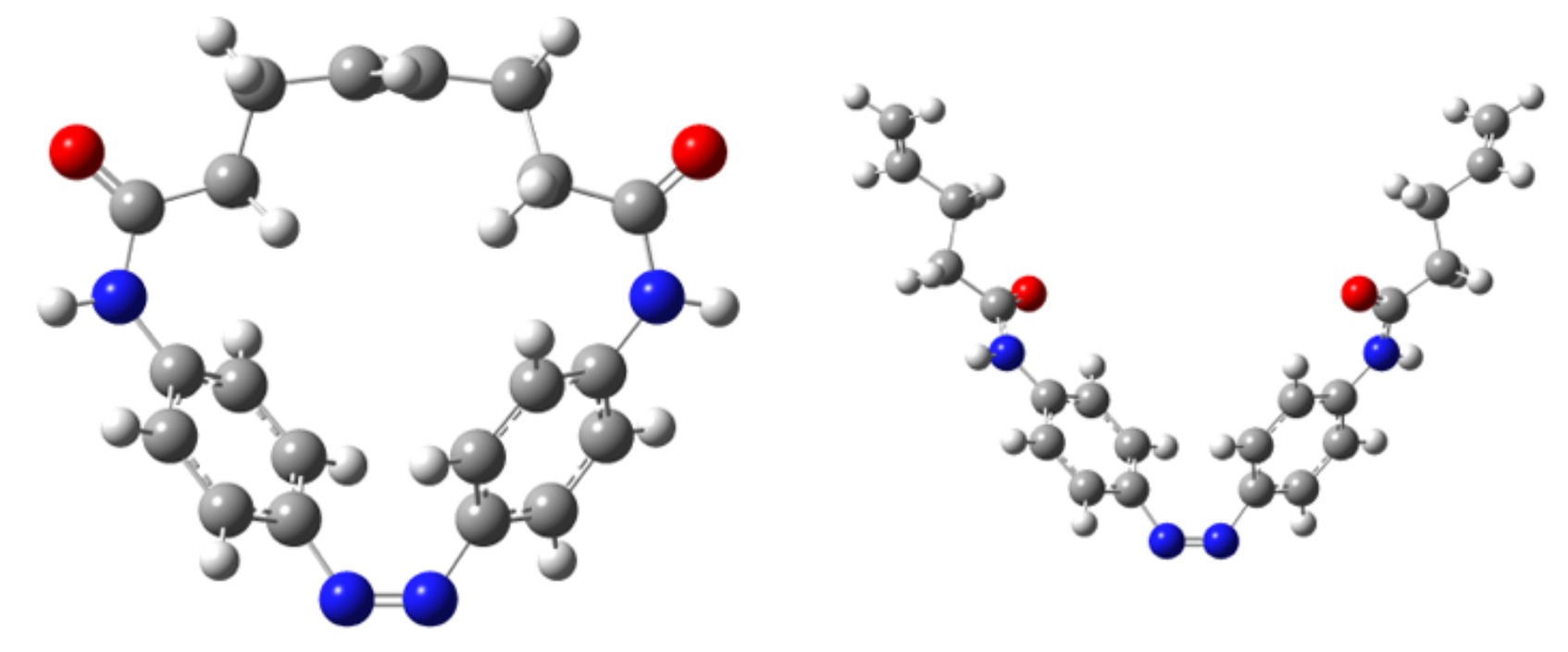
Photo-responsive polymers, including azobenzene (AB) and its derivatives have a wide range of potential applications as photo switches, molecular motors, and in optoelectronic devices. Separately, ring-opening metathesis polymerization (ROMP) is a powerful and widely used method for the controlled synthesis of polymer films. ROMP is a type of chain-growth polymerization where “ring” monomers open in the presence of a catalyst and, when there is strain in the closed ring, link together to form growing polymer chains.
Working with experimental collaborators, the Beck Group has used density functional theory calculations to identify the optimum design for a novel, light-responsive ring monomer expected to allow spatial and temporal control of ROMP via light-mediated changes in ring strain energy. Calculations show that trans-to-cis and cis-to-trans photoisomerization of the a particular AB-containing ring molecule can be achieved with monochromatic green and blue light, respectively, and will tune ring strain to turn ROMP on and off. The AB(2,2) monomer identified by the Beck Group will allow precise, reversible, spatial and temporal light-mediated control of ROMP through AB photoisomerization, and has promising potential applications in the fabrication of patterned and/or responsive polymer materials.
Q. Zhou*, I. Fursule, B. Berron and M. J. Beck. “Toward spatiotemporally controlled synthesis of photoresponsive polymers: Computational design of azobenzene-containing monomers for light-mediated ROMP”, J. Phys. Chem. A, 120 (36) 7101-7111 (2016).
Catalytic properties of ceria nanoparticles
CeO2 (ceria) is widely applied as an oxidation/reduction catalyst in industrial, automotive, and biomedical applications. Previous studies have connected the catalytic and autocatalytic activity of ceria to the presence of O-vacancies and associated Ce 3+ ions. Significant efforts have been dedicated to producing well-dispersed nanometer-scale ceria particles that maximize surface-to-volume ratio and surface O-vacancy concentration. Despite this, efforts to date have not yielded a definitive understanding of the atomistic mechanisms underlying the high activity of ceria nanoparticles (CNPs) as catalysts, catalyst supports, and/or anti-oxidants and O-scavengers.
Recent calculations in the Beck group have shown that CNPs, in contrast to bulk ceria samples, are super-oxidized, and are terminated with chemically absorbed O2 and O3molecules. These results suggest the possiblity that catalytic mechanisms at ultrasmall CNPs may not depend on the the thermodynamics and kinetics of O-vacancies. Current efforts focus on exploring catalytic mechanisms and CNP structures relevant in catalysis for energy and applications as biological anti-oxidants.
E. A. Grulke, M. J. Beck, R. A. Yokel, J. M. Unrine, U. M. Graham, M. L. Hancock. “Surface-controlled dissolution rates: a case study of nanoceria in carboxylic acid solutions” Env. Sci.: Nano 6 (5), 1478-1492 (2019).
X. Huang* and M. J. Beck. “Metal-free low-temperature water-gas shift catalysis over small, hydroxylated ceria nanoparticles”, ACS Catal., 5, 6362- 6369 (2015).
X. Huang* and M. J. Beck. “Size-dependent appearance of intrinsic Oq x “activated oxygen” molecules on ceria nanoparticles”, Chem. Mater., 27, 5840- 5844 (2015).
X. Huang* and M. J. Beck. “Determining the Oxidation State of Small, Hydroxylated Metal-Oxide Nanoparticles with Infrared Absorption Spectroscopy”, Chem. Mater., 27, 2965-2972 (2015).
E. Grulke, K. Reed, M. Beck, X. Huang*, A. Cormack, and S. Seal. “Nanoceria: factors affecting its pro- and anti-oxidant properties”, Env. Sci.: Nano, 1, 429-444 (2014). Invited Critical Review. Cover Image, developed with R. Yokel and M. Hazard.
X. Huang* and M. J. Beck. “Surface structure of catalytically-active ceria nanoparticles”, Comput. Mater. Sci., 91, 122-133 (2014). 23. X. Huang*, B. Wang, E. A. Grulke and M. J. Beck.
X. Huang*, B. Wang, E. A. Grulke and M. J. Beck. “Toward tuning the surface functionalizaton of small ceria nanoparticles”, J. Chem. Phys., 140, 074703 (2014).
Displacement Damage Dynamics
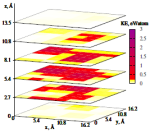
Understanding and modeling the structural damage produced in a target solid during ion implantation or irradiation in a space or fission/fusion environment is important in electronic device and energy applications. Using first-principles dynamical calculations we study displacement damage at the atomic-scale. Focusing on low-energy (<1 keV) displacements in Si and SiO2, we work to build a physical understanding of the connection between individual atomic displacements and any resulting stable defects.
Recent results have demonstrated the importance of dynamic disorder formation and relaxation and recrystallization processes in determining stable defect profiles. This dynamic disorder has been compared to the local melting behavior observed for higher energy displacements. Disorder is also generated along the displacement tracks of recoiling atoms. In SiO2 these disordered tracks can contribute to dielectric failure of gate oxides by providing a connected pathway of defect states within the oxide bandgap.
Ion Stopping Power from TDDFT

Time-dependent Density Functional Theory (TDDFT) rigorously extends density functional theory beyond the static, time-independent regime, allowing ab-initio calculation of excited states, optical properties and electron dynamics. A recent implementation of explicit timeintegration TDDFT in conjunction with MD for atomic nuclei (TDDFT+MD) allows direct, real-time calculation of the full quantum mechanical dynamics of many atom systems. This powerful method enables ab initio calculation of electron-mediated ion-solid interactions, electron-phonon coupling, electron transport in molecules and crystals, and driven catalytic processes.
Using this method we have studied the classic problem of the stopping power of well-channelled ions in Si. Using ions from across the periodic table we have calculated the so-called Z1 oscillations in stopping power with incident ion species. These oscillations were shown to arise from the fully dynamic interaction of the ion and target electrons, and quantitative agreement between experiments and calculation has been achieved without the use of free parameters.
Frenkel Pairs in Si
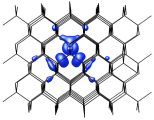
Point defects control the electronic properties of semiconductor materials and devices. The Si lattice vacancy and interstitial are generally accepted as prototype semiconductor point defects, and have been well characterized by both experimental and computational studies. Based on seminal EPR experiments dating to the 1960s, it was concluded that interstitials in Si migrate athermally preventing the appearance of stable vacancy-interstitial pairs, or Frenkel pairs (FPs).
Using DFT calculations of structures, total energies and charge densities, we have shown that charge redistribution between nearby interstitials and vacancies interferes with the athermal migration of the individual defects, suppressing FP recombination.
Ge Surface Properties
The self-assembly of nanostructures with novel electronic and optical properties has led to intense interest in understanding and controlling the heteroepitaxial growth and morphology of crystalline thin films. Heteroepitaxial Ge on Si(100) has served as a model system for many theoretical and experimental investigations in this area. Using DFT calculations we have studied surface and interface properties in the Ge/Si quantum dot system. We studied the wetting layer thickness and strain dependence of the surface and interface excess energy of a Ge layer atop a Si (001) substrate, and the surface excess energy of dimer vacancy line (DVL) reconstructed Ge (001) surfaces and the RS reconstructed Ge (105) surface as a function of applied strain.
Results of this work have elucidated the complex relationship between surface energy and strain energy in the formation of Ge quantum dots on Si (001), pointed to the strong impact of DVLs on strain in the Ge surface, and allowed for the determination of useful fits of surface excess energy versus strain for the Ge (001) and (105) surfaces from first principles.
D. Scopece*, M. J. Beck. “Epilayer thickness and strain dependence of Ge (113) surface energies”, Phys. Rev. B, 85, 155310 (2013).
D. Scopece*, F. Montalenti, and M. J. Beck. “Stability of Ge on Si (1 1 10) surfaces and the role of dimer tilting”, Phys. Rev. B, 85, 085312 (2012).
G. Chen, B. Sanduijav, D. Matei, G. Springholz, D. Scopece*, M. J. Beck, F. Montalenti, and L. Miglio. “Formation of Ge nanoripples on vicinal Si (1 1 10): From Stranski-Krastanow seeds to a perfectly faceted wetting layer”, Phys. Rev. Lett., 108, 055503 (2012).
M. Brehm, F. Montalenti, M. Grydlik, G. Vastola, H. Lichtenberger, N. Hrauda, M. J. Beck, T. Fromherz, F. Schaeffler, L. Miglio and G. Bauer. “Key role of the wetting layer in revealing the hidden path of Ge/Si(001) Stranski-Krastanow growth onset”, Phys. Rev. B, 80, 205321 (2009).
C. J. Moore, C. M. Retford, M. J. Beck, M. Asta, M. J. Miksis, P. W. Voorhees. “Orientation dependence of strained-Ge surface energies near (001): Role of dimer-vacancy lines and their interactions with steps”, Phys. Rev. Lett., 96, 126101 (2006).
O. E. Shklyaev, M. J. Beck, M. Asta, M. J. Miksis, and P. W. Voorhees. “Role of strain-dependent surface energies in Ge/Si (100) island formation”, Phys. Rev. Lett., 94, 176102 (2005).
M. J. Beck, A. van de Walle, and M. Asta. “Surface energetics and structure of the Ge wetting layer on Si (100)”, Phys. Rev. B, 70, 205337 (2004).
Learn More…
- Capabilities Summary and Research Interests
- Research Group Overview
- Kentucky Random Structures Toolkit (KRaSTk)
- Nanoscale Surface Properties
- Summary of Past Projects